Почему сгенерированная консенсусная последовательность vcf2fq пропускает отступы, даже если они преобладают? Как исправить?
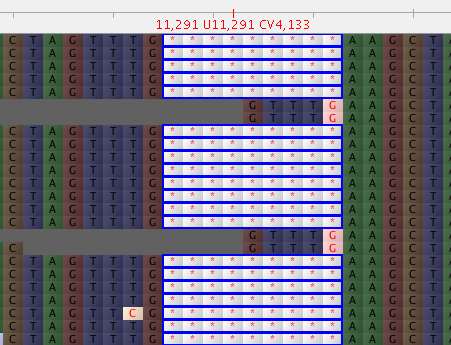
После выравнивания считываний последовательностей и преобразования в BAM я могу визуализировать существование делеции из 9 оснований.
Эта область удаления также правильно вызывается mpileup и bcftools.
bcftools mpileup -Ou -f $ref xxx.bam -o newbcfMPILE_xxx
bcftools call newbcfMPILE_xxx --ploidy 1 -mv -Ov -o newbcfMPILE_xxx_haploid.vcf
bcftools call newbcfMPILE_${sname} --ploidy 1 -c -Ov | vcfutils vcf2fq > cns_xxx.fq
В согласованной последовательности эта часть:
ctagtttgtctAgtttGaagcta <--consensus from vcf2fq
ctagtttg---------aagcta <--Expect this because reads with deletions is predominant
...........A....G...... <--mutations in other reads without deletion, which fill in the gaps in the consensus
ctagtttgtctGgtttTaagcta <--REF
В файле vcf я действительно вижу эти инделенные мутации с большим числом чтений с мутантными делециями, чем другие.
#CHROM POS REF ALT QUAL INFO
SARSCOV2 11287 GTCTGGTTTT G 228.344 DP=224; DP4=27,1,167,29;MQ=54
SARSCOV2 11288 TCTGGTTTTA T 228.325 DP=205; DP4=15,4,159,27;MQ=54
167 + 29 = 196 считываний из 224 показывают удаление. Другие делеции перекрываются, за исключением одного основания на обоих концах, с аналогичной доминирующей пропорцией.
Есть ли способ достичь консенсуса с удаленной частью, удаленной (или заполненной ---------) вместо нуклеотидов из числа прочтений меньшинства?