Как свернуть ветки в филогенетическом дереве по метке в их узлах или листьях?
Я построил филогенетическое древо для семейства белков, которое можно разделить на разные группы, классифицируя каждую по типу рецептора или типу ответа. Узлы в дереве помечены как тип рецептора.
В филогенетическом древе я вижу, что белки, принадлежащие к одним и тем же группам или типу рецепторов, сгруппированы вместе в одних и тех же ветвях. Поэтому я хотел бы свернуть эти ветви, которые имеют общие метки, сгруппировав их по заданному списку ключевых слов.
Команда будет выглядеть примерно так:
./collapse_tree_by_label -f phylogenetic_tree.newick -l list_of_labels_to_collapse.txt -o collapsed_tree.eps (или pdf)
Мой list_of_labels_to_collapse.txt будет выглядеть так: A B C D
Мое новое дерево было бы таким: (A_1:0,05,A_2:0,03,A_3:0,2,A_4:0,1):0,9,(((B_1:0,05,B_2:0,02,B_3:0,04):0,6 (C_1:0,6,C_2:0,08):0,7):0,5,(D_1:0,3,D_2:0,4,D_3:0,5,D_4:0,7,D_5:0.4):1.2)
Выходное изображение без свертывания выглядит так:
Свертывание выходного изображения должно быть таким (collapsed_tree.eps): 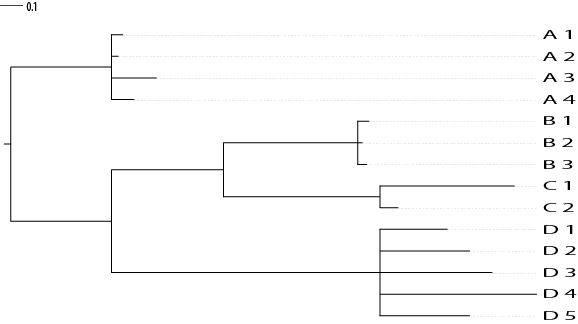
Ширина треугольников должна представлять длину ветви, а максимум треугольников должен представлять количество узлов в ветви.
Я играл с пакетом "ape" в R. Мне удалось построить филогенетическое дерево, но я до сих пор не могу понять, как свернуть ветви по ключевым словам в ярлыках:
require("ape")
Это загрузит дерево:
cat("((A_1:0.05,A_2:0.03,A_3:0.2,A_4:0.1):0.9,(((B_1:0.05,B_2:0.02,B_3:0.04):0.6,(C_1:0.6,C_2:0.08):0.7):0.5,(D_1:0.3,D_2:0.4,D_3:0.5,D_4:0.7,D_5:0.4):1.2):0.5);", file = "ex.tre", sep = "\n")
tree.test <- read.tree("ex.tre")
Здесь должен быть код, чтобы свернуть
Это построит дерево:
plot(tree.test)
5 ответов
Ваше дерево в том виде, в котором оно хранится в R, уже содержит подсказки, хранящиеся в виде политомий Это просто вопрос построения дерева с треугольниками, представляющими политомии.
Там нет функции в ape чтобы сделать это, что я знаю, но если вы немного возитесь с функцией печати, вы можете осуществить это
# Step 1: make edges for descendent nodes invisible in plot:
groups <- c("A", "B", "C", "D")
group_edges <- numeric(0)
for(group in groups){
group_edges <- c(group_edges,getMRCA(tree.test,tree.test$tip.label[grepl(group, tree.test$tip.label)]))
}
edge.width <- rep(1, nrow(tree.test$edge))
edge.width[tree.test$edge[,1] %in% group_edges ] <- 0
# Step 2: plot the tree with the hidden edges
plot(tree.test, show.tip.label = F, edge.width = edge.width)
# Step 3: add triangles
add_polytomy_triangle <- function(phy, group){
root <- length(phy$tip.label)+1
group_node_labels <- phy$tip.label[grepl(group, phy$tip.label)]
group_nodes <- which(phy$tip.label %in% group_node_labels)
group_mrca <- getMRCA(phy,group_nodes)
tip_coord1 <- c(dist.nodes(phy)[root, group_nodes[1]], group_nodes[1])
tip_coord2 <- c(dist.nodes(phy)[root, group_nodes[1]], group_nodes[length(group_nodes)])
node_coord <- c(dist.nodes(phy)[root, group_mrca], mean(c(tip_coord1[2], tip_coord2[2])))
xcoords <- c(tip_coord1[1], tip_coord2[1], node_coord[1])
ycoords <- c(tip_coord1[2], tip_coord2[2], node_coord[2])
polygon(xcoords, ycoords)
}
Тогда вам просто нужно пройтись по группам, чтобы добавить треугольники
for(group in groups){
add_polytomy_triangle(tree.test, group)
}
Я также искал этот вид инструмента целую вечность, не столько для свертывания категориальных групп, сколько для свертывания внутренних узлов на основе числового значения поддержки.
Функция di2multi в пакете ape может сворачивать узлы до политомий, но в настоящее время она может делать это только по порогу длины ветви. Вот грубая адаптация, которая позволяет вместо этого сворачиваться порогом значения поддержки узла (порог по умолчанию = 0,5).
Используйте на свой страх и риск, но у меня это работает на моем укоренившемся байесовском дереве.
di2multi4node <- function (phy, tol = 0.5)
# Adapted di2multi function from the ape package to plot polytomies
# based on numeric node support values
# (di2multi does this based on edge lengths)
# Needs adjustment for unrooted trees as currently skips the first edge
{
if (is.null(phy$edge.length))
stop("the tree has no branch length")
if (is.na(as.numeric(phy$node.label[2])))
stop("node labels can't be converted to numeric values")
if (is.null(phy$node.label))
stop("the tree has no node labels")
ind <- which(phy$edge[, 2] > length(phy$tip.label))[as.numeric(phy$node.label[2:length(phy$node.label)]) < tol]
n <- length(ind)
if (!n)
return(phy)
foo <- function(ancestor, des2del) {
wh <- which(phy$edge[, 1] == des2del)
for (k in wh) {
if (phy$edge[k, 2] %in% node2del)
foo(ancestor, phy$edge[k, 2])
else phy$edge[k, 1] <<- ancestor
}
}
node2del <- phy$edge[ind, 2]
anc <- phy$edge[ind, 1]
for (i in 1:n) {
if (anc[i] %in% node2del)
next
foo(anc[i], node2del[i])
}
phy$edge <- phy$edge[-ind, ]
phy$edge.length <- phy$edge.length[-ind]
phy$Nnode <- phy$Nnode - n
sel <- phy$edge > min(node2del)
for (i in which(sel)) phy$edge[i] <- phy$edge[i] - sum(node2del <
phy$edge[i])
if (!is.null(phy$node.label))
phy$node.label <- phy$node.label[-(node2del - length(phy$tip.label))]
phy
}
Это мой ответ, основанный на phytools::phylo.toBackbone функцию, см. http://blog.phytools.org/2013/09/even-more-on-plotting-subtrees-as.html и http://blog.phytools.org/2013/10/finding-edge-lengths-of-all-terminal.html. Сначала загрузите функцию в конце кода.
library(ape)
library(phytools) #phylo.toBackbone
library(phangorn)
cat("((A_1:0.05,E_2:0.03,A_3:0.2,A_4:0.1,A_5:0.1,A_6:0.1,A_7:0.35,A_8:0.4,A_9:01,A_10:0.2):0.9,((((B_1:0.05,B_2:0.05):0.5,B_3:0.02,B_4:0.04):0.6,(C_1:0.6,C_2:0.08):0.7):0.5,(D_1:0.3,D_2:0.4,D_3:0.5,D_4:0.7,D_5:0.4):1.2):0.5);", file = "ex.tre", sep = "\n")
phy <- read.tree("ex.tre")
groups <- c("A", "B|C", "D")
backboneoftree<-makebackbone(groups,phy)
# tip.label clade.label N depth
# 1 A_1 A 10 0.2481818
# 2 B_1 B|C 6 0.9400000
# 3 D_1 D 5 0.4600000
par(mfrow=c(2,2), mar=c(0,2,2,0) )
plot(phy, main="Complete tree" )
plot(backboneoftree)
makebackbone<-function(groupings,phy){
listofspecies<-phy$tip.label
listtopreserve<-list()
lengthofclades<-list()
meandistnode<-list()
newedgelengths<-list()
for (i in 1:length(groupings)){
groupings<-groups
bestmrca<-getMRCA(phy,grep(groupings[i], phy$tip.label) )
mrcatips<-phy$tip.label[unlist(phangorn::Descendants(phy,bestmrca, type="tips") )]
listtopreserve[i]<- mrcatips[1]
meandistnode[i]<- mean(dist.nodes(phy)[unlist(lapply(mrcatips,
function(x) grep(x, phy$tip.label) ) ),bestmrca] )
lengthofclades[i]<-length(mrcatips)
provtree<-drop.tip(phy,mrcatips, trim.internal=F, subtree = T)
n3<-length(provtree$tip.label)
newedgelengths[i]<-setNames(provtree$edge.length[sapply(1:n3,function(x,y)
which(y==x),y=provtree$edge[,2])],
provtree$tip.label)[provtree$tip.label[grep("tips",provtree$tip.label)] ]
}
newtree<-drop.tip(phy,setdiff(listofspecies,unlist(listtopreserve)),
trim.internal = T)
n<-length(newtree$tip.label)
newtree$edge.length[sapply(1:n,function(x,y)
which(y==x),y=newtree$edge[,2])]<-unlist(newedgelengths)+unlist(meandistnode)
trans<-data.frame(tip.label=newtree$tip.label,clade.label=groupings,
N=unlist(lengthofclades), depth=unlist(meandistnode) )
rownames(trans)<-NULL
print(trans)
backboneoftree<-phytools::phylo.toBackbone(newtree,trans)
return(backboneoftree)
}

РЕДАКТИРОВАТЬ: я не пробовал это, но это может быть другой ответ: "Сценарий и функция для преобразования ветвей кончика дерева, то есть толщины или треугольников, с шириной обоих коррелирует с определенными параметрами (например, номер вида клана) (tip.branches.R) " http://www.sysbot.biologie.uni-muenchen.de/en/people/cusimano/use_r.html http://www.sysbot.biologie.uni-muenchen.de/en/people/cusimano/tip.branches.R
Я думаю, что сценарий, наконец, делает то, что я хотел. Исходя из ответа, который дал @CactusWoman, я немного изменил код, чтобы скрипт попытался найти MRCA, представляющую наибольшую ветвь, соответствующую моему шаблону поиска. Это решило проблему не слияния неполитомных ветвей или свертывания всего дерева, потому что один соответствующий узел был ошибочно вне правильной ветви.
Кроме того, я включил параметр, который представляет ограничение для отношения количества шаблонов в данной ветви, чтобы мы могли выбирать и сворачивать / группировать ветви, у которых, например, по крайней мере 90% советов соответствуют шаблону поиска.
library(geiger)
library(phylobase)
library(ape)
#functions
find_best_mrca <- function(phy, group, threshold){
group_matches <- phy$tip.label[grepl(group, phy$tip.label, ignore.case=TRUE)]
group_mrca <- getMRCA(phy,phy$tip.label[grepl(group, phy$tip.label, ignore.case=TRUE)])
group_leaves <- tips(phy, group_mrca)
match_ratio <- length(group_matches)/length(group_leaves)
if( match_ratio < threshold){
#start searching for children nodes that have more than 95% of descendants matching to the search pattern
mrca_children <- descendants(as(phy,"phylo4"), group_mrca, type="all")
i <- 1
new_ratios <- NULL
nleaves <- NULL
names(mrca_children) <- NULL
for(new_mrca in mrca_children){
child_leaves <- tips(tree.test, new_mrca)
child_matches <- grep(group, child_leaves, ignore.case=TRUE)
new_ratios[i] <- length(child_matches)/length(child_leaves)
nleaves[i] <- length(tips(phy, new_mrca))
i <- i+1
}
match_result <- data.frame(mrca_children, new_ratios, nleaves)
match_result_sorted <- match_result[order(-match_result$nleaves,match_result$new_ratios),]
found <- numeric(0);
print(match_result_sorted)
for(line in 1:nrow(match_result_sorted)){
if(match_result_sorted$ new_ratios[line]>=threshold){
return(match_result_sorted$mrca_children[line])
found <- 1
}
}
if(found==0){return(found)}
}else{return(group_mrca)}
}
add_triangle <- function(phy, group,phylo_plot){
group_node_labels <- phy$tip.label[grepl(group, phy$tip.label)]
group_mrca <- getMRCA(phy,group_node_labels)
group_nodes <- descendants(as(tree.test,"phylo4"), group_mrca, type="tips")
names(group_nodes) <- NULL
x<-phylo_plot$xx
y<-phylo_plot$yy
x1 <- max(x[group_nodes])
x2 <-max(x[group_nodes])
x3 <- x[group_mrca]
y1 <- min(y[group_nodes])
y2 <- max(y[group_nodes])
y3 <- y[group_mrca]
xcoords <- c(x1,x2,x3)
ycoords <- c(y1,y2,y3)
polygon(xcoords, ycoords)
return(c(x2,y3))
}
#main
cat("((A_1:0.05,E_2:0.03,A_3:0.2,A_4:0.1,A_5:0.1,A_6:0.1,A_7:0.35,A_8:0.4,A_9:01,A_10:0.2):0.9,((((B_1:0.05,B_2:0.05):0.5,B_3:0.02,B_4:0.04):0.6,(C_1:0.6,C_2:0.08):0.7):0.5,(D_1:0.3,D_2:0.4,D_3:0.5,D_4:0.7,D_5:0.4):1.2):0.5);", file = "ex.tre", sep = "\n")
tree.test <- read.tree("ex.tre")
# Step 1: Find the best MRCA that matches to the keywords or search patten
groups <- c("A", "B|C", "D")
group_labels <- groups
group_edges <- numeric(0)
edge.width <- rep(1, nrow(tree.test$edge))
count <- 1
for(group in groups){
best_mrca <- find_best_mrca(tree.test, group, 0.90)
group_leaves <- tips(tree.test, best_mrca)
groups[count] <- paste(group_leaves, collapse="|")
group_edges <- c(group_edges,best_mrca)
#Step2: Remove the edges of the branches that will be collapsed, so they become invisible
edge.width[tree.test$edge[,1] %in% c(group_edges[count],descendants(as(tree.test,"phylo4"), group_edges[count], type="all")) ] <- 0
count = count +1
}
#Step 3: plot the tree hiding the branches that will be collapsed/grouped
last_plot.phylo <- plot(tree.test, show.tip.label = F, edge.width = edge.width)
#And save a copy of the plot so we can extract the xy coordinates of the nodes
#To get the x & y coordinates of a plotted tree created using plot.phylo
#or plotTree, we can steal from inside tiplabels:
last_phylo_plot<-get("last_plot.phylo",envir=.PlotPhyloEnv)
#Step 4: Add triangles and labels to the collapsed nodes
for(i in 1:length(groups)){
text_coords <- add_triangle(tree.test, groups[i],last_phylo_plot)
text(text_coords[1],text_coords[2],labels=group_labels[i], pos=4)
}
Это не относится к изображению кладов в виде треугольников, но помогает с коллапсом узлов с низкой поддержкой. Библиотекаggtreeимеет функциюas.polytomyкоторый можно использовать для свертывания узлов на основе значений поддержки.
Например, чтобы свернуть бутстрапы менее чем на 50%, вы должны использовать:
polytree = as.polytomy(raxtree, feature='node.label', fun=function(x) as.numeric(x) < 50)