umap выделяет две разные модели
Я пытаюсь создать umap для данных одной ячейки из образцов человека и образцов ptx. Я могу получить umap туда, где он показывает umap с различными кластерами, но я хочу показать, где находятся образцы ptx и где находятся образцы людей.
Мой код выглядит следующим образом:
#create the Seurat object
OD_10K_HUMAN <- CreateSeuratObject(counts = HUMAN_OD_10K.data, min.cells = 0, project = "human")
SD_5K_HUMAN <- CreateSeuratObject(counts = HUMAN_SD_5K.data, min.cells = 0, project = "human")
BNL.5K <- CreateSeuratObject(counts = SD_BNL_5K.data, min.cells = 0, project = "ptx")
BNL.10K <- CreateSeuratObject(counts = OD_BNL_10K.data, min.cells = 0, project = "ptx")
BNM.10K <- CreateSeuratObject(counts = OD_BNM_10K.data, min.cells = 0, project = "ptx")
BNM.5K <- CreateSeuratObject(counts = SD_BNM_5K.data, min.cells = 0, project = "ptx")
#merge data
scData <- merge(BNL.10K, y = c(BNL.5K, BNM.10K, BNM.5K, SD_5K_HUMAN, OD_10K_HUMAN), add.cell.ids = c("A", "B", "C", "D", "E", "F"), project = "HTB2876")
mark the mito genes
mito.genes <- grep(pattern = "^MT-", x = rownames(x = scData), value = TRUE)
length(mito.genes)
scData[["percent.mt"]] <- PercentageFeatureSet(scData, pattern = "^MT-")
scData[["log_nCount_RNA"]] <- log2(scData[["nCount_RNA"]]+1)
# remove cells with <200 RNA molecules, or >6000 molecules, or >30% mito
scData <- subset(scData, subset = nFeature_RNA > 200 & nFeature_RNA < 6000 & percent.mt < 30)
scData <- NormalizeData(scData, normalization.method = "LogNormalize", scale.factor = 10000)
scData <- FindVariableFeatures(scData, selection.method = "vst", nfeatures = 2000)
all.genes <- rownames(scData)
scData <- ScaleData(scData, features = VariableFeatures(object = scData), vars.to.regress = c("nCount_RNA"))
scData <- RunPCA(scData, features = VariableFeatures(object = scData))
#DimPlot(scData, reduction = "pca")
numPC = 20
scData <- FindNeighbors(scData, dims = 1:numPC)
scData <- FindClusters(scData, resolution = 0.4)
scData <- RunUMAP(scData, dims = 1:numPC)
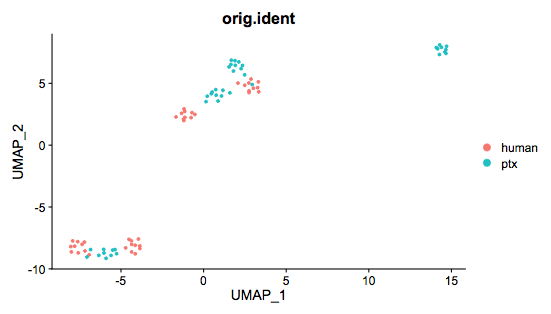
DimPlot(scData, reduction = "umap", label = TRUE)
1 ответ
Должен быть столбец
orig.ident в ваших метаданных, поэтому предположим, что мы объединяем данные, как вы:
library(Seurat)
library(magrittr)
data1 = CreateSeuratObject(counts = pbmc_small[["RNA"]]@counts[,1:40],project="human")
data2 = CreateSeuratObject(counts = pbmc_small[["RNA"]]@counts[,41:80],project="ptx")
scData <- merge(data1,data2)
Запускаем umap:
scData = scData %>%
SCTransform() %>%
RunPCA() %>%
RunUMAP(dims=1:15)
Участок:
DimPlot(scData,group.by="orig.ident")