Раскраска линий кривой разрежения метаданными (пакет vegan) (пакет phyloseq)
Первый раз задаешь вопрос здесь. Я не смог найти ответ на этот вопрос в других постах (love stackexchange, кстати).
Во всяком случае... Я создаю кривую разрежения через веганский пакет, и я получаю очень грязный график, который имеет очень толстую черную полосу в нижней части графика, которая скрывает некоторые линии выборки с низким разнообразием. В идеале я хотел бы создать график со всеми моими линиями (169; я мог бы уменьшить его до 144), но создать составной график, раскрасив по образцу года и создавая разные типы линий для каждого пруда (т.е. 2 года образца: 2016, 2017 и 3 пруда: 1,2,5). Я использовал phyloseq для создания объекта со всеми моими данными, а затем разделил свою таблицу изобилия OTU из моих метаданных на отдельные объекты (jt = таблица OTU и sampledata = metadata). Мой текущий код:
jt <- as.data.frame(t(j)) # transform it to make it compatible with the proceeding commands
rarecurve(jt
, step = 100
, sample = 6000
, main = "Alpha Rarefaction Curve"
, cex = 0.2
, color = sampledata$PondYear)
# A very small subset of the sample metadata
Pond Year
F16.5.d.1.1.R2 5 2016
F17.1.D.6.1.R1 1 2017
F16.1.D15.1.R3 1 2016
F17.2.D00.1.R2 2 2017
2 ответа
Вот пример того, как построить кривую разрежения с помощью ggplot. Я использовал данные, доступные в пакете phyloseq, доступном от bioconductor.
установить phyloseq:
source('http://bioconductor.org/biocLite.R')
biocLite('phyloseq')
library(phyloseq)
нужны другие библиотеки
library(tidyverse)
library(vegan)
данные:
mothlist <- system.file("extdata", "esophagus.fn.list.gz", package = "phyloseq")
mothgroup <- system.file("extdata", "esophagus.good.groups.gz", package = "phyloseq")
mothtree <- system.file("extdata", "esophagus.tree.gz", package = "phyloseq")
cutoff <- "0.10"
esophman <- import_mothur(mothlist, mothgroup, mothtree, cutoff)
извлечь таблицу OTU, транспонировать и преобразовать во фрейм данных
otu <- otu_table(esophman)
otu <- as.data.frame(t(otu))
sample_names <- rownames(otu)
out <- rarecurve(otu, step = 5, sample = 6000, label = T)
Теперь у вас есть список, каждый элемент соответствует одному образцу:
Очистите список немного:
rare <- lapply(out, function(x){
b <- as.data.frame(x)
b <- data.frame(OTU = b[,1], raw.read = rownames(b))
b$raw.read <- as.numeric(gsub("N", "", b$raw.read))
return(b)
})
список меток
names(rare) <- sample_names
преобразовать во фрейм данных:
rare <- map_dfr(rare, function(x){
z <- data.frame(x)
return(z)
}, .id = "sample")
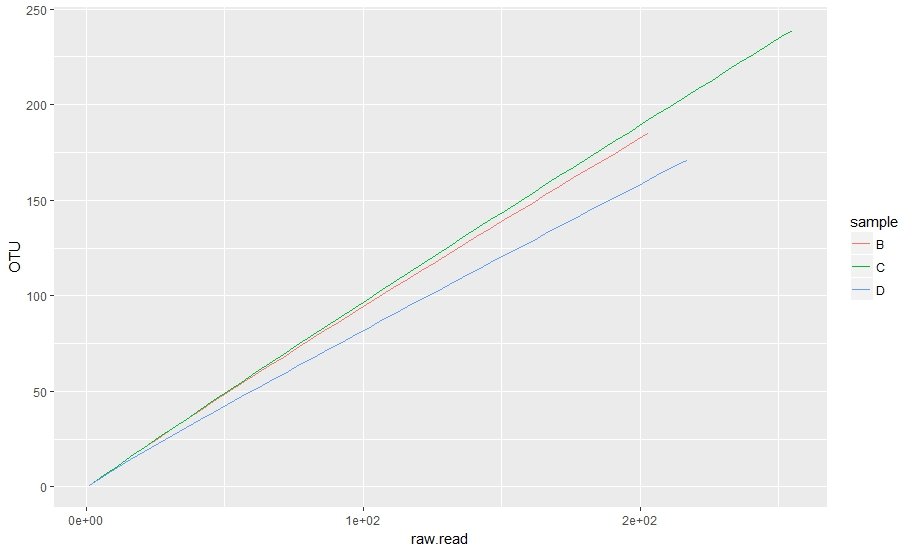
Давайте посмотрим, как это выглядит:
head(rare)
sample OTU raw.read
1 B 1.000000 1
2 B 5.977595 6
3 B 10.919090 11
4 B 15.826125 16
5 B 20.700279 21
6 B 25.543070 26
сюжет с ggplot2
ggplot(data = rare)+
geom_line(aes(x = raw.read, y = OTU, color = sample))+
scale_x_continuous(labels = scales::scientific_format())
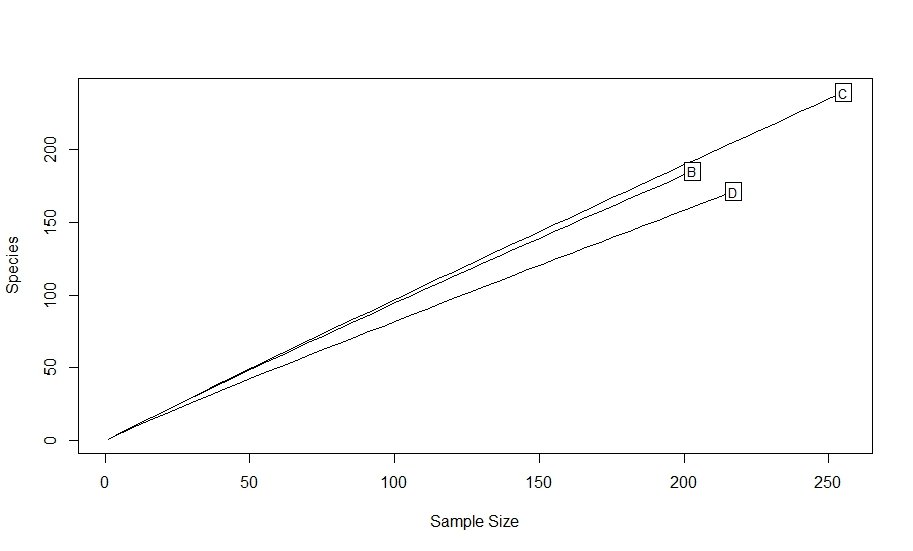
веганский сюжет:
rarecurve(otu, step = 5, sample = 6000, label = T) #low step size because of low abundance
Можно сделать дополнительный столбец групп и цвета в соответствии с этим.
Вот пример, как добавить другую группировку. Предположим, у вас есть таблица вида:
groupings <- data.frame(sample = c("B", "C", "D"),
location = c("one", "one", "two"), stringsAsFactors = F)
groupings
sample location
1 B one
2 C one
3 D two
где образцы сгруппированы согласно другой функции. Вы могли бы использовать lapply или же map_dfr перейти groupings$sample и этикетка rare$location,
rare <- map_dfr(groupings$sample, function(x){ #loop over samples
z <- rare[rare$sample == x,] #subset rare according to sample
loc <- groupings$location[groupings$sample == x] #subset groupings according to sample, if more than one grouping repeat for all
z <- data.frame(z, loc) #make a new data frame with the subsets
return(z)
})
head(rare)
sample OTU raw.read loc
1 B 1.000000 1 one
2 B 5.977595 6 one
3 B 10.919090 11 one
4 B 15.826125 16 one
5 B 20.700279 21 one
6 B 25.543070 26 one
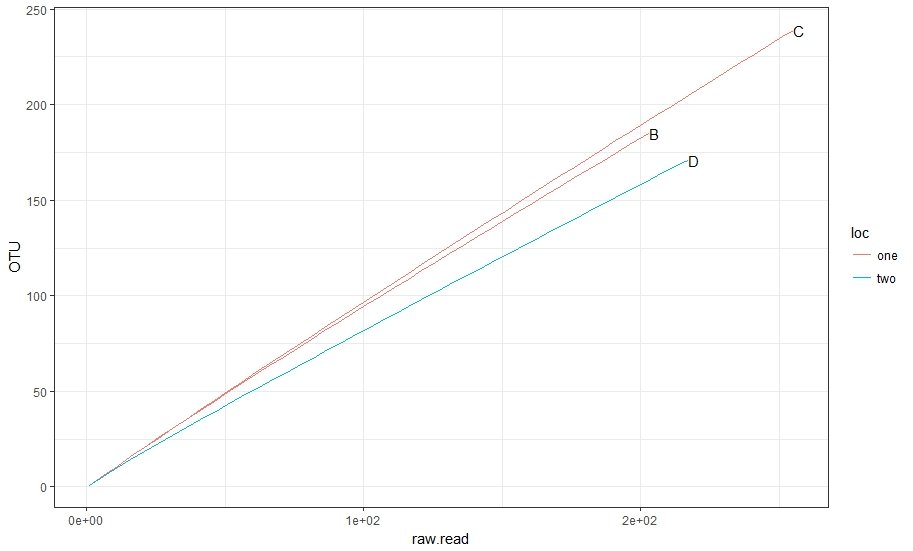
Давайте сделаем из этого приличный сюжет
ggplot(data = rare)+
geom_line(aes(x = raw.read, y = OTU, group = sample, color = loc))+
geom_text(data = rare %>% #here we need coordinates of the labels
group_by(sample) %>% #first group by samples
summarise(max_OTU = max(OTU), #find max OTU
max_raw = max(raw.read)), #find max raw read
aes(x = max_raw, y = max_OTU, label = sample), check_overlap = T, hjust = 0)+
scale_x_continuous(labels = scales::scientific_format())+
theme_bw()
Я знаю, что это старый вопрос, но я изначально пришел сюда по той же причине и по пути узнал, что в недавнем обновлении (2021 г.) это стало НАМНОГО проще.
Это абсолютно голый пример.
В конечном счете, мы собираемся отображать окончательный результат в ggplot, чтобы у вас были полные возможности настройки, и это решение tidyverse с dplyr.
library(vegan)
library(dplyr)
library(ggplot2)
Я собираюсь использовать данные о дюнах вveganи сгенерировать столбец случайных метаданных для сайта.
data(dune)
metadata <- data.frame("Site" = as.factor(1:20),
"Vegetation" = rep(c("Cactus", "None")))
Сейчас мы запустим, но предоставьте аргументtidy = TRUEкоторый будет экспортировать фрейм данных, а не график.
Здесь следует отметить, что я также использовалstepаргумент. Шаг по умолчанию равен 1, и это означает, что по умолчанию вы получите одну строку на человека для каждой выборки в вашем наборе данных, что может сделать результирующий фрейм данных огромным. Шаг = 1 дляduneдал мне более 600 строк. Слишком большое уменьшение шага сделает ваши кривые блочными, так что это будет баланс между шагом и разрешением для хорошего сюжета.
Затем я передал левое соединение прямо вrarecurveвызов
dune_rare <- rarecurve(dune,
step = 2,
tidy = TRUE) %>%
left_join(metadata)
Теперь он будет отображаться в ggplot с вызовом цвета/цвета для любых метаданных, которые вы прикрепили. Отсюда вы можете настроить и другие аспекты сюжета.
ggplot(dune_rare) +
geom_line(aes(x = Sample, y = Species, group = Site, colour = Vegetation)) +
theme_bw()
(Извините, мне пока не разрешено вставлять изображение :()