R: круговой сюжет - как построить несвязанные области между секторами с минимальным перекрытием
У меня есть фрейм данных с общими чертами между 4 группами пациентов и типами клеток. У меня много разных функций, но общих (присутствующих в более чем 1 группе) - это всего несколько.
Я хочу сделать обходной сюжет, который отражает несколько связей между общими функциями между группами пациентов и типами ячеек, и в то же время дает представление о том, сколько нет общих функций в каждой группе.
На мой взгляд, это должен быть график с четырьмя секторами (по одному для каждой группы пациентов и типов клеток) с несколькими связями между ними. Размер каждого сектора должен отражать общее количество объектов в группе, и большая часть этой области не должна быть связана с другими группами, а должна быть пустой.
Это то, что у меня есть, но я не хочу секторов, выделенных для каждой функции, только для каждой группы пациентов и типов клеток.
MWE:
library(circlize)
patients <- c(rep("patient1",20), rep("patient2",10))
cell.types <- c(rep("cell1",12), rep("cell2",8),rep("cell1",6), rep("cell2",4))
features <- c(paste("feature",1:12,sep="_"), paste("feature",9:16,sep="_"), paste("feature",c(1,2,9,10,17,18),sep="_"), paste("feature",c(1,18,19,20),sep="_"))
dat <- data.frame(patient=patients, cell.type=cell.types, feature=features)
dat
dat <- with(dat, table(paste(patient,cell.type,sep='|'), feature))
dat
chordDiagram(as.data.frame(dat), transparency = 0.5)

РЕДАКТИРОВАТЬ!!
То, что @m-dz показывает в своем ответе, - это на самом деле формат, который я ищу, 4 сектора для 4 различных комбинаций типа "пациент / клетка", показывающие только соединения, в то время как несвязанные функции, хотя и не показаны, должны учитывать размер сектора.
Тем не менее, я понимаю, что у меня есть более сложный сценарий, чем в MWE выше.
Считается, что функция появляется в 2 группах пациента / типа ячейки, не только когда она идентична в 2 группах, но также и когда она похожа... (идентичность последовательности выше порога). Таким образом, у меня есть увольнения...
Функция A в Patient1-Cell1 может быть подключена к функции A в Patient2-Cell1, но также и к функции B... Функция A должна учитываться только один раз (уникальный счет) для пациента 1-Cell1 и расширяться до 2 различных функций в Patient2-cell1.
Ниже приведен пример того, как мои фактические данные выглядят более точно, и посмотрите, если работать с этим примером, мы сможем получить окончательный цирковой сюжет! Спасибо!!
##MWE
#NON OVERLAPPING SETS!
#1: non-shared features
nonshared <- data.frame(patient=c(rep("pat1",20), rep("pat2",10)), cell.type=c(rep("cell1",12), rep("cell2",8),rep("cell1",6), rep("cell2",4)), feature=paste("a",1:30,sep=''))
nonshared
#2: features shared between cell types within same patient
sharedcells <- data.frame(patient=c(rep("pat1",3), rep("pat2",4)), cell.types=c(rep("cell1||cell2",3),rep("cell1||cell2",4)), features=c("b1||b1","b1||b1","b1||b1","b2||b2","b3||b3","b4||b4","b4||b5"))
sharedcells
#3: features shared between patients within same cell types
sharedpats <- data.frame(patients=c(rep("pat1||pat2",2), rep("pat1||pat2",6)), cell.type=c(rep("cell1",2),rep("cell2",6)), features=c("c1||c1","c2||c1","c3||c3","c3||c4","c3||c5","c6||c5","c7||c7","c8||c8"))
sharedpats
#4: features shared between patients and cell types
#4.1: shared across pat1-cell1, pat1-cell2, pat2-cell1, pat2-cell2
sharedall1 <- data.frame(both=c(rep("pat1-cell1||pat1-cell2||pat2-cell1||pat2-cell2",4)), features=c("d1||d1||d1||d1","d2||d2||d2||d3","d4||d4||d3||d3","d5||d5||d5||d5"))
#4.2: shared across pat1-cell1, pat1-cell2, pat2-cell1
sharedall2 <- data.frame(both=c(rep("pat1-cell1||pat1-cell2||pat2-cell1",2)), features=c("d6||d6||d6","d7||d7||d7"))
#4.3: shared across pat1-cell1, pat1-cell2, pat2-cell2
sharedall3 <- data.frame(both="pat1-cell1||pat1-cell2||pat2-cell2", features="d8||d8||d9")
#4.4: shared across pat1-cell1, pat2-cell1, pat2-cell2
sharedall4 <- data.frame(both="pat1-cell1||pat2-cell1||pat2-cell2", features="d10||d10||d9")
#4.5: shared across pat1-cell2, pat2-cell1, pat2-cell2
sharedall5 <- data.frame(both=c(rep("pat1-cell2||pat2-cell1||pat2-cell2",3)), features=c("d11||d11||d11","d12||d13||d13","d12||d14||d14"))
#4.6: shared across pat1-cell1, pat2-cell2
sharedall6 <- data.frame()
#4.7: shared across pat1-cell2, pat2-cell1
sharedall7 <- data.frame(both=c(rep("pat1-cell2||pat2-cell1",2)), features=c("d15||d16","d17||d17"))
sharedall <- rbind(sharedall1, sharedall2, sharedall3, sharedall4, sharedall5, sharedall6, sharedall7)
sharedall
#you see there might be overlaps between the different subsets of sharedall, but not between sharedall, sharedparts, sharedcells, and nonshared
#I NEED A CIRCOS PLOT THAT SHOWS ALL THE CONNECTIONS. THE NON-CONNECTED (nonshared) FEATURES SHOULD NOT BE SHOWN, BUT THE SHOULD COUNT TO THE SIZE OF THE SECTOR (CORRESPONDING TO A PATIENT-CELL COMBINATION)
#THE FEATURES SHOULD BE COUNT UNIQUELY, SO IF THERE ARE ENTRIES LIKE:
#3 pat1||pat2 cell2 c3||c3
#4 pat1||pat2 cell2 c3||c4
#5 pat1||pat2 cell2 c3||c5
#THE FEATURE c3 SHOULD BE COUNT ONCE FOR pat1, AND EXPAND TO 3 DIFFERENT FEATURES IN pat2
3 ответа
Дополнительное примечание об ожидаемом результате: цель состояла в том, чтобы создать график, показывающий просто, сколько объектов совместно используется, игнорируя отдельные функции (1-й график ниже) или общие функции перекрываются (например, на 2-м графике похоже, что одни и те же объекты совместно используются всеми групп, что неверно, если смотреть на 1-й сюжет, но здесь важно соотношение функций, общих для групп).
Приведенный ниже код дает следующие две цифры (рис. 1 оставлен для справки):
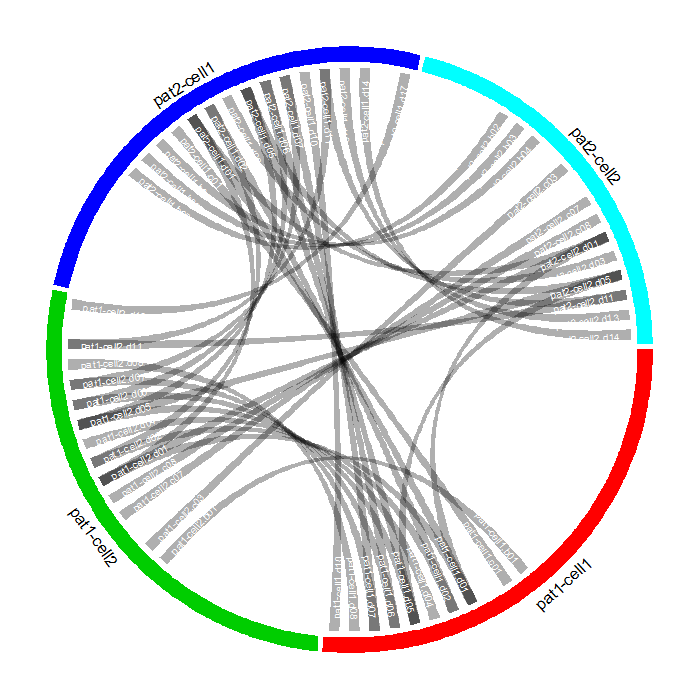
Все индивидуальные особенности
Простой подсчет уникальных и общих функций
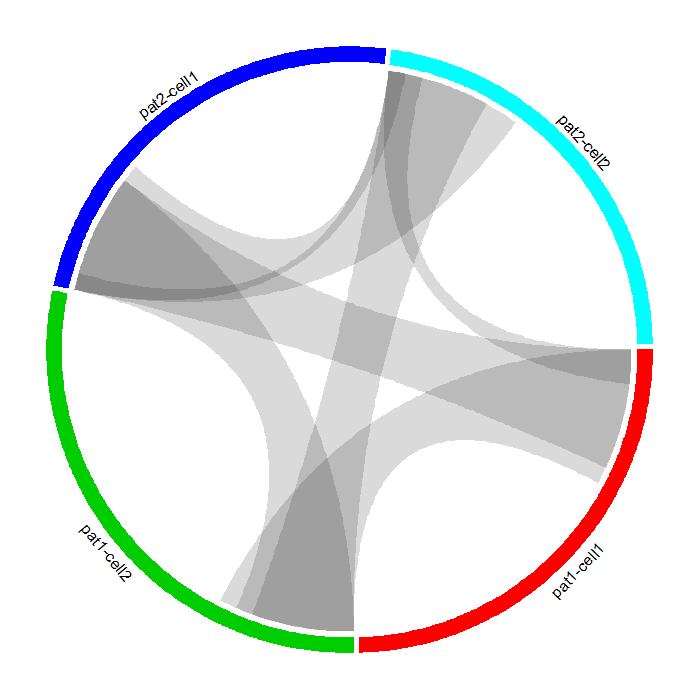
Один из них должен соответствовать ожиданиям.
# Prep. data --------------------------------------------------------------
nonshared <- data.frame(patient=c(rep("pat1",20), rep("pat2",10)), cell.type=c(rep("cell1",12), rep("cell2",8),rep("cell1",6), rep("cell2",4)), feature=paste("a",1:30,sep=''))
sharedcells <- data.frame(patient=c(rep("pat1",3), rep("pat2",4)), cell.types=c(rep("cell1||cell2",3),rep("cell1||cell2",4)), features=c("b1||b1","b1||b1","b1||b1","b2||b2","b3||b3","b4||b4","b4||b5"))
sharedpats <- data.frame(patients=c(rep("pat1||pat2",2), rep("pat1||pat2",6)), cell.type=c(rep("cell1",2),rep("cell2",6)), features=c("c1||c1","c2||c1","c3||c3","c3||c4","c3||c5","c6||c5","c7||c7","c8||c8"))
sharedall1 <- data.frame(both=c(rep("pat1-cell1||pat1-cell2||pat2-cell1||pat2-cell2",4)), features=c("d1||d1||d1||d1","d2||d2||d2||d3","d4||d4||d3||d3","d5||d5||d5||d5"))
sharedall2 <- data.frame(both=c(rep("pat1-cell1||pat1-cell2||pat2-cell1",2)), features=c("d6||d6||d6","d7||d7||d7"))
sharedall3 <- data.frame(both="pat1-cell1||pat1-cell2||pat2-cell2", features="d8||d8||d9")
sharedall4 <- data.frame(both="pat1-cell1||pat2-cell1||pat2-cell2", features="d10||d10||d9")
sharedall5 <- data.frame(both=c(rep("pat1-cell2||pat2-cell1||pat2-cell2",3)), features=c("d11||d11||d11","d12||d13||d13","d12||d14||d14"))
sharedall6 <- data.frame()
sharedall7 <- data.frame(both=c(rep("pat1-cell2||pat2-cell1",2)), features=c("d15||d16","d17||d17"))
sharedall <- rbind(sharedall1, sharedall2, sharedall3, sharedall4, sharedall5, sharedall6, sharedall7)
#I NEED A CIRCOS PLOT THAT SHOWS ALL THE CONNECTIONS. THE NON-CONNECTED (nonshared) FEATURES SHOULD NOT BE SHOWN, BUT THE SHOULD COUNT TO THE SIZE OF THE SECTOR (CORRESPONDING TO A PATIENT-CELL COMBINATION)
#THE FEATURES SHOULD BE COUNT UNIQUELY, SO IF THERE ARE ENTRIES LIKE:
#3 pat1||pat2 cell2 c3||c3
#4 pat1||pat2 cell2 c3||c4
#5 pat1||pat2 cell2 c3||c5
#THE FEATURE c3 SHOULD BE COUNT ONCE FOR pat1, AND EXPAND TO 3 DIFFERENT FEATURES IN pat2
# Start -------------------------------------------------------------------
library(circlize)
library(data.table)
library(magrittr)
library(stringr)
library(RColorBrewer)
# Split and pad with 0 ----------------------------------------------------
fun <- function(x) unlist(tstrsplit(x, split = '||', fixed = TRUE))
nonshared %>% setDT()
sharedcells %>% setDT()
sharedpats %>% setDT()
sharedall %>% setDT()
nonshared <- nonshared[, .(group = paste(patient, cell.type, sep = '-'), feature)][, feature := paste0('a', str_pad(str_extract(feature, '[0-9]+'), 2, 'left', '0'))]
sharedcells <- sharedcells[, lapply(.SD, fun), by = 1:nrow(sharedcells)][, .(group = paste(patient, cell.types, sep = '-'), feature = features)][, feature := paste0('b', str_pad(str_extract(feature, '[0-9]+'), 2, 'left', '0'))]
sharedpats <- sharedpats[, lapply(.SD, fun), by = 1:nrow(sharedpats)][, .(group = paste(patients, cell.type, sep = '-'), feature = features)][, feature := paste0('c', str_pad(str_extract(feature, '[0-9]+'), 2, 'left', '0'))]
sharedall <- sharedall[, lapply(.SD, fun), by = 1:nrow(sharedall)][, .(group = both, feature = features)][, feature := paste0('d', str_pad(str_extract(feature, '[0-9]+'), 2, 'left', '0'))]
dt_split <- rbindlist(
list(
nonshared,
sharedcells,
sharedpats,
sharedall
)
)
# Set key and self join to find shared features ---------------------------
setkey(dt_split, feature)
dt_join <- dt_split[dt_split, .(group, i.group, feature), allow.cartesian = TRUE] %>%
.[group != i.group, ]
# Create a "sorted key" ---------------------------------------------------
# key := paste(sort(.SD)...
# To leave only unique combinations of groups and features
dt_join <-
dt_join[,
key := paste(sort(.SD), collapse = '|'),
by = 1:nrow(dt_join),
.SDcols = c('group', 'i.group')
] %>%
setorder(feature, key) %>%
unique(by = c('key', 'feature')) %>%
.[, .(
group_from = i.group,
group_to = group,
feature = feature)]
# Rename and key ----------------------------------------------------------
dt_split %>% setnames(old = 'group', new = 'group_from') %>% setkey(group_from, feature)
dt_join %>% setkey(group_from, feature)
# Individual features -----------------------------------------------------
# Features without connections --------------------------------------------
dt_singles <- dt_split[, .(group_from, group_to = group_from, feature)] %>%
.[, N := .N, by = feature] %>%
.[!(N > 1 & group_from == group_to), !c('N')]
# Bind all, add some columns etc. -----------------------------------------
dt_bind <- rbind(dt_singles, dt_join) %>% setorder(group_from, feature, group_to)
dt_bind[, ':='(
group_from_f = paste(group_from, feature, sep = '.'),
group_to_f = paste(group_to, feature, sep = '.'))]
dt_bind[, feature := NULL] # feature can be removed
# Colour
dt_bind[, colour := ifelse(group_from_f == group_to_f, "#FFFFFF00", '#00000050')] # Change first to #FF0000FF to show red blobs
# Prep. sectors -----------------------------------------------------------
sectors_f <- union(dt_bind[, group_from_f], dt_bind[, group_to_f]) %>% sort()
colour_lookup <-
union(dt_bind[, group_from], dt_bind[, group_to]) %>% sort() %>%
structure(seq_along(.) + 1, names = .)
sector_colours <- str_replace_all(sectors_f, '.[a-d][0-9]+', '') %>%
colour_lookup[.]
# Gaps between sectors ----------------------------------------------------
gap_sizes <- c(0.0, 1.0)
gap_degree <-
sapply(table(names(sector_colours)), function(i) c(rep(gap_sizes[1], i-1), gap_sizes[2])) %>%
unlist() %>% unname()
# gap_degree <- rep(0, length(sectors_f)) # Or no gap
# Plot! -------------------------------------------------------------------
# Each "sector" is a separate patient/cell/feature combination
circos.par(gap.degree = gap_degree)
circos.initialize(sectors_f, xlim = c(0, 1))
circos.trackPlotRegion(ylim = c(0, 1), track.height = 0.05, bg.col = sector_colours, bg.border = NA)
for(i in 1:nrow(dt_bind)) {
row_i <- dt_bind[i, ]
circos.link(
row_i[['group_from_f']], c(0, 1),
row_i[['group_to_f']], c(0, 1),
border = NA, col = row_i[['colour']]
)
}
# "Feature" labels
circos.trackPlotRegion(track.index = 2, ylim = c(0, 1), panel.fun = function(x, y) {
sector.index = get.cell.meta.data("sector.index")
circos.text(0.5, 0.25, sector.index, col = "white", cex = 0.6, facing = "clockwise", niceFacing = TRUE)
}, bg.border = NA)
# "Patient/cell" labels
for(s in names(colour_lookup)) {
sectors <- sectors_f %>% { .[str_detect(., s)] }
highlight.sector(
sector.index = sectors, track.index = 1, col = colour_lookup[s],
text = s, text.vjust = -1, niceFacing = TRUE)
}
circos.clear()
# counts of unique and shared features ------------------------------------
xlims <- dt_split[, .N, by = group_from][, .(x_from = 0, x_to = N)] %>% as.matrix()
links <- dt_join[, .N, by = .(group_from, group_to)]
colours <- dt_split[, unique(group_from)] %>% structure(seq_along(.) + 1, names = .)
library(circlize)
sectors = names(colours)
circos.par(cell.padding = c(0, 0, 0, 0))
circos.initialize(sectors, xlim = xlims)
circos.trackPlotRegion(ylim = c(0, 1), track.height = 0.05, bg.col = colours, bg.border = NA)
for(i in 1:nrow(links)) {
link <- links[i, ]
circos.link(link[[1]], c(0, link[[3]]), link[[2]], c(0, link[[3]]), col = '#00000025', border = NA)
}
# "Patient/cell" labels
for(s in sectors) {
highlight.sector(
sector.index = s, track.index = 1, col = colours[s],
text = s, text.vjust = -1, niceFacing = TRUE)
}
circos.clear()
Изменить: Просто добавив ссылку из удаленного комментария: Смотрите этот ответ для хорошего примера маркировки!
@ M-DZ обеспечивает правильное направление. Я могу предоставить более подробную информацию о ваших смоделированных данных.
Давайте начнем отсюда:
patients <- c(rep("patient1",20), rep("patient2",10))
cell.types <- c(rep("cell1",12), rep("cell2",8),rep("cell1",6), rep("cell2",4))
features <- c(paste("feature",1:12,sep="_"), paste("feature",9:16,sep="_"), paste("feature",c(1,2,9,10,17,18),sep="_"), paste("feature",c(1,18,19,20),sep="_"))
dat <- data.frame(patient=patients, cell.type=cell.types, feature=features)
dat <- with(dat, table(paste(patient,cell.type,sep='|'), feature))
as.data.frame новообращенные dat к фрейму данных с тремя столбцами (то есть список смежности, где ссылки начинаются с первого столбца и указывают на второй столбец)
dat = as.data.frame(dat, stringsAsFactors = FALSE)
Генерация цветов для пациентов / клеток и функций.
features = unique(dat[[2]])
features_col = structure(rand_color(length(features)), names = features)
patients_col = structure(2:5, names = unique(dat[[1]]))
Если функция существует только в одной комбинации пациент / клетка, вы не хотите показывать ее, но хотите сохранить ее положение на графике, вы можете просто установить#FFFFFF00 как его цвет (белый цвет с полной прозрачностью, чтобы он не покрывал другие ссылки). Здесь мы хотим, чтобы цвет ссылки был таким же, как у секторов объектов.
col = ifelse(dat[[3]], features_col[dat[[2]]], "#FFFFFF00")
col = gsub("FF$", "80", col) # half transparent
features_count = tapply(dat[[3]], dat[[2]], sum)
# set color to white if it only exists in one patient/cell
col[features_count[dat[[2]]] == 1] = "#FFFFFF00"
И финальная диаграмма аккордов:
chordDiagram(dat, col = col, grid.col = c(features_col, patients_col))
Вы можете видеть, что в тематических секторах есть как минимум две ссылки, указывающие на пациентов / ячейки.

Подготовить данные
library(circlize)
patients <- c(rep("patient1",20), rep("patient2",10))
cell.types <- c(rep("cell1",12), rep("cell2",8),rep("cell1",6), rep("cell2",4))
features <- c(paste("feature",1:12,sep="_"), paste("feature",9:16,sep="_"), paste("feature",c(1,2,9,10,17,18),sep="_"), paste("feature",c(1,18,19,20),sep="_"))
dat <- data.frame(patient=patients, cell.type=cell.types, feature=features)
dat <- with(dat, table(paste(patient,cell.type,sep='|'), feature))
dat<-as.data.frame(dat,stringsAsFactors = FALSE)
получить все комбинации пациента и типов клеток
df=NULL
for(i in levels(as.factor(dat$feature))){
temp<-as.data.frame(matrix(combn(dat[which(dat$feature==i),1],2),byrow = TRUE,ncol=2),stringsAsFactors = FALSE)
temp$feature=i
temp$Freq=1
Freq_0<-subset(dat$Var1,dat$feature==i & dat$Freq==0)
for(j in Freq_0){
temp$Freq[temp$V1==j | temp$V2==j]=0
}
df<-rbind(df,temp)
}

добавить цвет
df$color=rainbow(dim(df)[1])
df[which(df$Freq==0),5]="white"
df$Freq=1
chordDiagram(df[,c(-3,-5)], transparency = 0.5,col = df$color)
другая ссылка означает другую функцию, а цвет ссылки белый, где 'Freq' равен 0 
Я превращаю цвет "белый" в "черный", в то время как черный более заметен 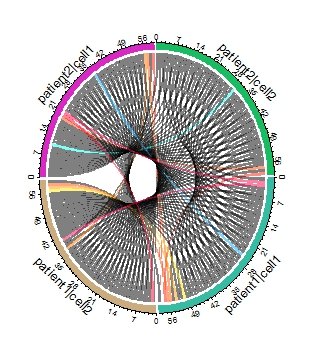
Если вы хотите оставить атрибут "функция"...... давайте сначала подготовим данные
library(circlize)
patients <- c(rep("patient1",20), rep("patient2",10))
cell.types <- c(rep("cell1",12), rep("cell2",8),rep("cell1",6), rep("cell2",4))
features <- c(paste("feature",1:12,sep="_"), paste("feature",9:16,sep="_"), paste("feature",c(1,2,9,10,17,18),sep="_"), paste("feature",c(1,18,19,20),sep="_"))
dat <- data.frame(patient=patients, cell.type=cell.types, feature=features)
dat <- with(dat, table(paste(patient,cell.type,sep='|'), feature))
dat<-as.data.frame(dat,stringsAsFactors = FALSE)
df=NULL
for(i in levels(as.factor(dat$feature))){
temp<-as.data.frame(matrix(combn(dat[which(dat$feature==i),1],2),byrow = TRUE,ncol=2),stringsAsFactors = FALSE)
temp$feature=i
temp$Freq=1
Freq_0<-subset(dat$Var1,dat$feature==i & dat$Freq==0)
for(j in Freq_0){
temp$Freq[temp$V1==j | temp$V2==j]=0
}
df<-rbind(df,temp)
}
обработал это
library(dplyr)
df1<-subset(df,df$Freq==1)
df0<-subset(df,df$Freq==0)
df1_mod<-summarise(group_by(df1,V1,V2),Freq=n())
df0_mod<-summarise(group_by(df0,V1,V2),Freq=n())
добавить цвет
df1_mod$color<-rainbow(5)
df0_mod$color<-"white"
df_res<-rbind(df0_mod,df1_mod)
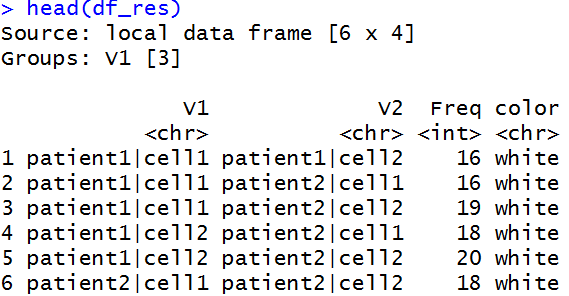
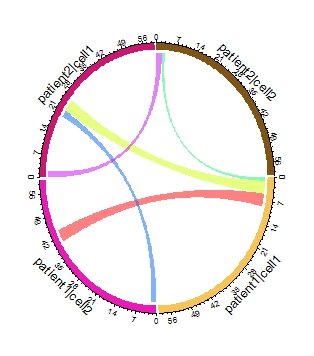
Нарисуй это
chordDiagram(df_res, transparency = 0.5,col = df_res$color)
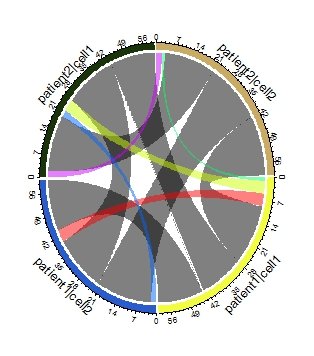
Это изображение показывает, что в "Freq" много нулей.