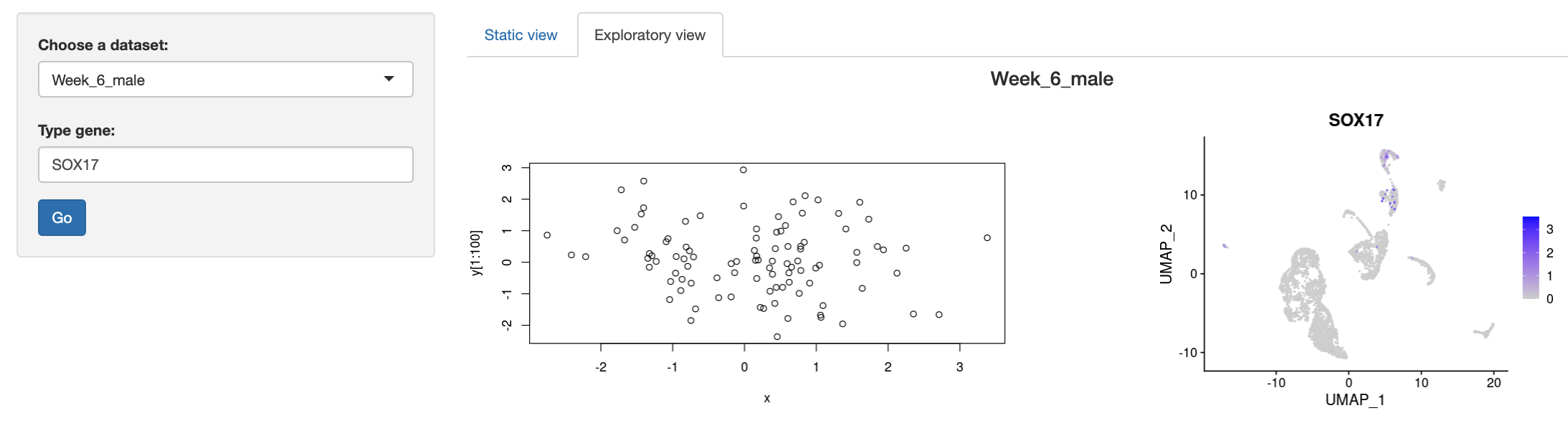
Не удается отобразить график в блестящем приложении на сервере, но работает на локальном сервере и на сервере Rstudio
Я застрял в этой проблеме несколько дней.
Я создаю блестящее приложение, в котором пользователь выбирает набор данных, а затем сервер читает соответствующий файл.rds и отображает графики. Для графического инструмента требуется пакет Seurat.
Когда я тестировал на местном уровне, он работает. Затем я развертываю на блестящем сервере, расположенном в нашем университете, приложения также хорошо работают на сервере Rstudio.
Но когда я подключаюсь через внешний источник, не отображаются графики, в которых говорится: "Произошла ошибка. Проверьте свои журналы или свяжитесь с автором приложения для уточнения".
Я погуглил и нашел возможные причины, связанные с окружающей средой:
- путь к файлу Я это проверял, и, поскольку я тестировал на том же компьютере (сервер Rstudio такой же, как блестящий сервер), это может не быть проблемой?
- package Когда я загружаю библиотеку (Seurat) в начале моего блестящего app.R, внешняя ссылка не работает, но сервер Rstudio работает хорошо. Итак, я попробовал ggplot2, чтобы проверить, является ли это проблемой пакета, он загружается без проблем и графики отображаются.
Чтобы быть конкретным, похоже, что это проблема совместимости Seurat, но почему на той же машине он работает на сервере Rstudio? Сообщество Seurat заявило, что эта проблема была решена в новом выпуске CRAN (3.2.0), но почему я все еще сталкиваюсь с этой проблемой? Как исправить..??
Это то, что я заметил, и надеюсь, что вы, эксперты, дадите мне некоторое представление...
Вот код для чтения.rds и отображения графиков:
# read .rds
germ <- readRDS(file.path(paste("/srv/shiny-server/GermlinAtlas/data/",input$dataset,".rds", sep="")))
# render plot
observeEvent(input$dataset, {
if (is.null(input$dataset)) return(NULL)
if (is.null(input$gene)) return(NULL)
infile <- isolate(input$dataset)
germ <- readRDS(file.path(paste("/srv/shiny-server/GermlinAtlas/data/",infile,".rds", sep=""))) # read .rds
gene <- isolate(input$gene)
x <- rnorm(100) # for ggplot test
y <- rnorm(300) # for ggplot test
output$dim <- renderPlot({
DimPlot(germ, reduction = "umap") # plot with Seurat
plot(x,y[1:100]) # plot with ggplot2
})
output$featureplot <- renderPlot({
FeaturePlot(germ, input$gene) # plot with Seurat
})
})
вывод с сервера Rstudio:
вывод с сервера (со стороны пользователя):