PyMOL получить название выбранного белка
Поэтому я использую скрипт PyMOL для поиска поверхностных остатков в белке (находится по адресу http://www.pymolwiki.org/index.php/FindSurfaceResidues). И мне нужно написать текстовый файл, содержащий название белка, который в данный момент выбран в сеансе PyMOL. Насколько я искал, я не могу найти команду PyMOL, чтобы получить имя выбранного белка. Кто-нибудь с большим опытом работы с PyMOL имеет представление о том, как это сделать?
Спасибо
2 ответа
Не уверен, что получаю вопрос ОП, верно, измененный код, позволяющий записать файл с именем обнаруженного белка/объекта, на которомfindSurfaceResidues()сталкивается со списком остатков, составляющих поверхность, и файлом, в котором перечислены все атомы, входящие в состав поверхности. Мне пришлось изменить часть doShow исходного сценария, потому что я не мог передать выделенные фрагменты вfindSurfaceResiduesкоманда.
from __future__ import print_function
from pymol import cmd, stored
def my_save(file,object,header):
# make sure there's a newline after the header
suff = "" if header[-1]=='\n' else '\n'
f = open(file, 'w')
# write the header
f.write("################\n##"+ header+suff+"################\n")
# write the PDB
f.write(cmd.get_pdbstr(object))
f.close()
def findSurfaceAtoms(selection = "all", cutoff = 2.5, quiet = 1):
"""
DESCRIPTION
Finds those atoms on the surface of a protein
that have at least 'cutoff' exposed A**2 surface area.
USAGE
findSurfaceAtoms [ selection, [ cutoff ]]
SEE ALSO
findSurfaceResidues
"""
#REMOVE WATERS HOH
cmd.select('all_waters', 'resn HOH')
cmd.remove('all_waters')
cutoff, quiet = float(cutoff), int(quiet)
tmpObj = cmd.get_unused_name("_tmp")
cmd.create(tmpObj, "(" + selection + ") and polymer", zoom=0)
cmd.set("dot_solvent", 1, tmpObj)
cmd.get_area(selection=tmpObj, load_b=1)
# threshold on what one considers an "exposed" atom (in A**2):
cmd.remove(tmpObj + " and b < " + str(cutoff))
selName = cmd.get_unused_name("exposed_atm_")
cmd.select(selName, "(" + selection + ") in " + tmpObj)
cmd.delete(tmpObj)
if not quiet:
print("Exposed atoms are selected in: " + selName)
print("cmd.get_names('objects', 1 ,selection) : ", cmd.get_names('objects', 1 ,selection))
print("cmd.get_names(, 1 ,selection) : ", cmd.get_names('selections', 1 ,selection))
print('selName : ', selName , type(selName))
return selName
def findSurfaceResidues(selection="all", cutoff=2.5, doShow=0, quiet=1):
"""
DESCRIPTION
Finds those residues on the surface of a protein
that have at least 'cutoff' exposed A**2 surface area.
USAGE
findSurfaceResidues [ selection, [ cutoff, [ doShow ]]]
ARGUMENTS
selection = string: object or selection in which to find exposed
residues {default: all}
cutoff = float: cutoff of what is exposed or not {default: 2.5 Ang**2}
RETURNS
(list: (chain, resv ) )
A Python list of residue numbers corresponding
to those residues w/more exposure than the cutoff.
"""
cutoff, doShow, quiet = float(cutoff), int(doShow), int(quiet)
selName = findSurfaceAtoms(selection, cutoff, quiet)
exposed = set()
cmd.iterate(selName, "exposed.add((chain,resv))", space=locals())
selNameRes = cmd.get_unused_name("exposed_res_")
cmd.select(selNameRes, "byres " + selName)
if not quiet:
print("Exposed residues are selected in: " + selNameRes)
if doShow:
print('------------------DO SHOW -------')
# cmd.show_as("spheres", "(" + selection + ") and polymer")
cmd.show_as("spheres", selection + " and polymer")
cmd.color("white", selection)
cmd.color("yellow", selNameRes)
cmd.color("red", selName)
print_locals(selNameRes)
return sorted(exposed)
cmd.extend("findSurfaceAtoms", findSurfaceAtoms)
cmd.extend("findSurfaceResidues", findSurfaceResidues)
def print_locals(sele):
"""
SALVA TUTTI I RESIDUI CHE COMPONGONO LA SELZIONE
SAVE NAMES OF ALL RESIDUES IN THE SELECTION
"""
stored.residues = dict()
print('---------------------> ', cmd.get_names('objects', 1 , sele))
for _name in cmd.get_names('objects', 1 , sele):
stored.residues[_name] = []
#CMD.ITERATE 1
cmd.iterate(sele, "stored.residues[model].append((resv, resn))")
#CMD.ITERATE 2
# cmd.iterate(sele, "stored.residues[model].append((resv, resn, [type, index, name]))") ## !!!!!!!!!!!!!!!!!!!!! FARE QJUI
# print(stored.residues)
## THIS BLOCK WORKS ONLY WITH CMD.ITERATE 1
for _i in stored.residues:
b = list(dict.fromkeys(stored.residues.get(_i)))
stored.residues[_i] = b
with open('surfaces.txt', 'w') as _handle:
for _i in stored.residues:
_handle.write(_i+'\n')
# CMD.ITERATE 1
_handle.writelines([str(_a[0]).ljust(6) + str(_a[1].rjust(10))+'\n' for _a in stored.residues[_i]])
#CMD.ITERATE 2
# _handle.writelines([str(_a[0]).ljust(6) + str(_a[1].rjust(10)) + ' '.join(str(item) for item in [i for i in _a[2]]).rjust(40)+ '\n' for _a in stored.residues[_i]])
_handle.write('------------------------------------------------------\n')
#SAVE PDB CONTAINING SURFACE RESIDUES :
#SAVE PDB CONTAINING SURFACE RESIDUES (see my_saves above from https://sourceforge.net/p/pymol/mailman/message/29103774/:
my_save('surface.pdb' , sele, ' '.join([_i for _i in stored.residues]))
cmd.extend("print_locals", print_locals)
если вы загружаете, например2lyz.pdbв pymol и запустите скрипт, вызвав:
findSurfaceResidues(doShow=1, quiet =0)покажет:
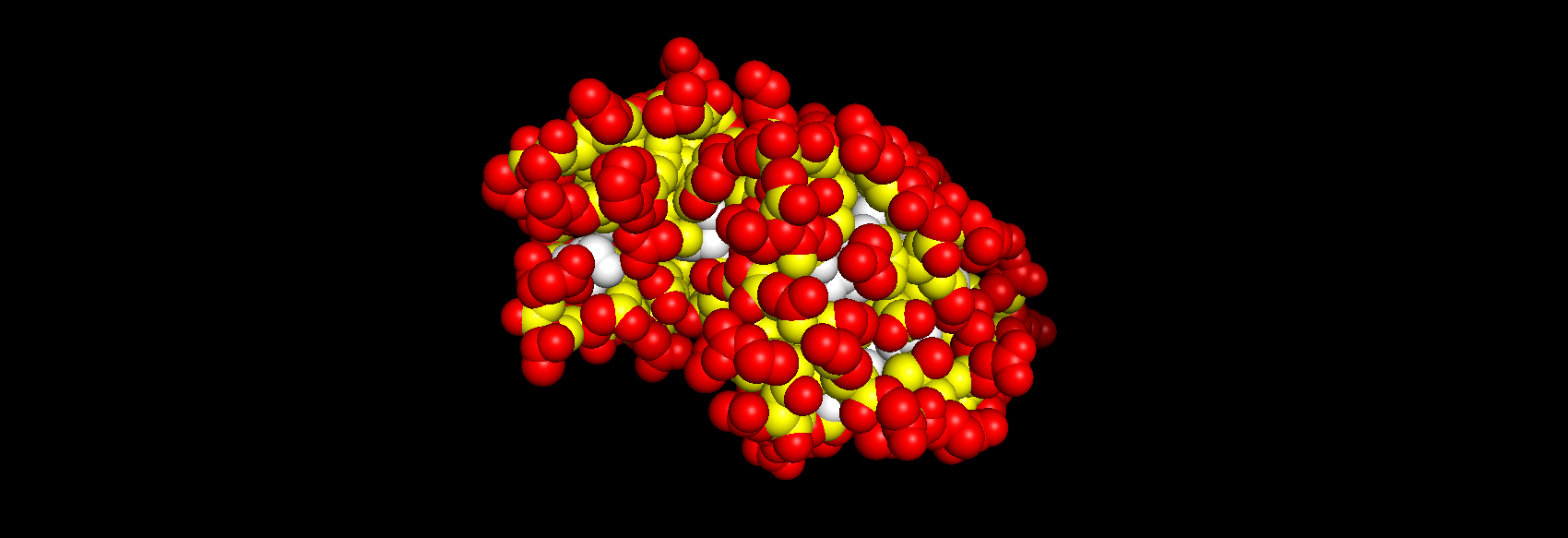
и сохранит файлsurface.txt:
2lyz
1 LYS
2 VAL
3 PHE
4 GLY
5 ARG
6 CYS
7 GLU
10 ALA
11 ALA
12 MET
13 LYS
14 ARG
.............
и файлsurface.pdb:
################
##2lyz
################
ATOM 1 N LYS A 1 3.246 10.041 10.379 1.00 5.28 N
ATOM 2 CA LYS A 1 2.386 10.407 9.247 1.00 7.90 C
ATOM 3 C LYS A 1 2.462 11.927 9.098 1.00 7.93 C
ATOM 4 O LYS A 1 2.582 12.668 10.097 1.00 6.28 O
ATOM 5 CB LYS A 1 0.946 9.964 9.482 1.00 3.54 C
ATOM 6 CG LYS A 1 -0.045 10.455 8.444 1.00 3.75 C
ATOM 7 CD LYS A 1 -1.470 10.062 8.818 1.00 2.85 C
ATOM 8 CE LYS A 1 -2.354 9.922 7.589 1.00 3.83 C
ATOM 9 NZ LYS A 1 -3.681 9.377 7.952 1.00 1.78 N1+
ATOM 10 N VAL A 2 2.383 12.351 7.847 1.00 9.08 N
ATOM 11 CA VAL A 2 2.387 13.773 7.480 1.00 9.24 C
ATOM 12 C VAL A 2 0.998 14.050 6.903 1.00 6.36 C
.............